Research Priorities in The Ehlers-Danlos Syndromes and Hypermobility Spectrum Disorders
INTRODUCTION
The Ehlers-Danlos syndromes (EDS) and hypermobility spectrum disorders (HSD) are complex conditions. There remain many unanswered questions. These questions include our understanding of the basic sciences at a genetic and protein level and their interactions with the environment; the functional changes in disease (pathophysiology) that arise from related disorders and their true relationships with EDS and HSD; the best ways to measure and treat symptoms related to EDS and HSD; the social impact of ill-health; and the best ways to educate and share information.
The Ehlers-Danlos Society has established a roadmap to develop and convey its research priorities, in collaboration with The International Consortium on EDS and HSD. We aim to inform researchers and funders of areas of interest that they may wish to explore or support.
These priorities are based The Society’s ongoing discussions with many stakeholders including the EDS and HSD community, our boards and coalitions, The International Consortium, researchers, industry leads, donors and grant awarding bodies. To ensure that the portfolio remains live, up to date, we encourage views from our whole community.
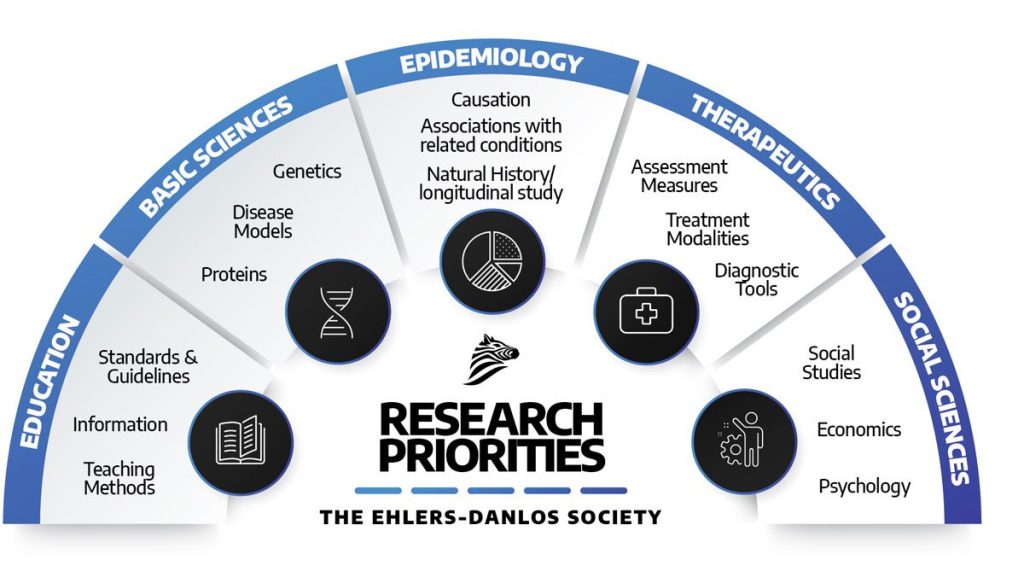
Our portfolio recognizes that there are a variety of disciplines within research. In cataloging the priorities, we have considered these in the context of:
- Basic Sciences (Genetics, molecular protein studies, disease models, i.e., how the disease develops and testing potential treatment approaches);
- Epidemiology (Study of the incidence and prevalence of diseases, association between conditions, causes of illness, natural history of illness);
- Therapeutics (Tools for diagnosis, tools for measuring the effects of treatment, treatments);
- Social Sciences (Social studies, health economics, the psychology of illness); and
- Education (Teaching methods, Information, Standards, and Guideline engagement).
Addressing these research priorities will lead to improving the lives of all those impacted by EDS and HSD, and will significantly lead to a better understanding for healthcare professionals, providers, and the economies it impacts.
We aspire to offer grants annually, with calls for clinical research proposals early in the year and for basic science later in the year. We also offer grants of varying value to reflect the different nature of researcher requirements. These include microgrants and support for two-to-three-year projects. Examples of our recent funding are available to see here.
As a researcher, please look out for announcements of grant-funding rounds on our website, here.
Genetic and molecular characterization
Despite revolutionary advances in genetic research and genomic techniques, there is still a substantial amount of EDS patients in whom current molecular testing fails to reveal the underlying genetic cause, leaving them without a definite diagnosis. This leads to a lack of recognition of their disorder, lack of accurate counseling possibilities, insufficient or inappropriate management strategies, and consequential distress and reduced quality of life.
Defining and diagnosing hEDS remains a challenge. Despite multiple attempts, no definitive molecular explanation has been found for people with this diagnosis. Several factors could contribute to this apparent failure, including lack of clarity for inclusion criteria for the diagnosis and locus heterogeneity (pathogenic variants in more than one gene are responsible for the phenotype). Furthermore, several factors play into the phenotypic presentation such as gender, age, training, pain threshold, and multiple genetic and non‐genetic factors. Large-scale international database registries of phenotypic traits, combined with large-scale genetic screening projects are necessary to uncover phenotypic patterns, that can help the design and interpretation of genetic data in the pursuit of the genetic etiology(ies) of hEDS.
In addition, some individuals present with EDS-phenotypes that do not fall within one of the currently recognized types of EDS, clinically nor genetically, implying that the genetic heterogeneity of EDS, other than hEDS, has also not been completely resolved.
Research priorities
- To assemble sufficient collections of biological material corresponding to families and/or cohorts (similar groups) of patients, whose phenotypic characteristics have been correctly analyzed. The collection of data and high-quality biological samples, as well as their storage and dissemination, are of fundamental importance. The development, consolidation, and sustainability of biobanks is a specific area requiring support.
- Mapping and cloning of the disease responsible genes; identification of pathogenic variants; detection of gene deletions or other anomalies of gene dosage or number of genes present.
Pathophysiology – The Function and Symptoms of a Disease (or of Types of EDS and HSD)
Over the past two decades, much progress has been made in identifying novel genes for distinct EDS phenotypes, introducing new and sometimes unexpected pathogenic concepts. This opens up unprecedented possibilities to study the pathological mechanisms underlying the diverse features seen in EDS. The identification of these genetic pathogenic variants should be followed by appropriate pathophysiological studies to allow the development of new therapeutic strategies. This research requires the use of different approaches. Several cellular and animal models that mirror the genetic defects and pathophysiological mechanisms of patients with different EDS types are currently available. Nonetheless, the study of animal models for EDS is still in its early stages, and creation of additional models reflecting different affected genes and different types of genetic variants, is needed. These models can be used to yield new insights into disease mechanisms, to identify clinically targetable biomarkers, to unravel signaling pathways and cellular processes for the development of personalized therapies, and to perform preclinical pharmacological studies.
Research priorities
- Development of transgenic animal and imaging facilities;
- Study of in vitro cell-based models
- Integrative analysis of ‘omics’ data, including genomics, transcriptomics, proteomics, matrisomics, metabolomics, to identify common and/or distinct pathophysiological mechanisms between the different EDS subtypes
- In-depth study of the collagen ultrastructure in different EDS types and HSD.
- Identification of the appropriate non-genetic markers, biological, functional etc., to be used for diagnosis, and evaluation of disease progression.
For most types of EDS and for HSD there is a relatively little natural history data. People with EDS or HSD do not always know what to expect at different stages in their lives. This applies equally to children and young people, and adults.
Clinical studies addressing prevalence and patterns of features impacting on quality of life among the different EDS types and HSD are needed, including the cardiovascular manifestations, chronic widespread pain, fatigue, and associated disorders such as structural and functional gastro-intestinal disorders, Postural Orthostatic Tachycardia Syndrome and other autonomic concerns, psychological health, and immune hypersensitivity.
Moreover, insights in genotype-phenotype correlations are currently generally insufficient to counsel affected individuals regarding the prognosis of their disease. The development of international registries, coupling detailed clinical data with genetic data, and the development of tools needed to implement these studies, in particular data management tools for shared databases linked to biobanks are needed.
Research priorities
- The collection of information on the different EDS types and HSD in terms of incidence and prevalence
- The definition of new categories or nosological entities through in-depth analysis, at clinical/genetic level, of apparently homogeneous or the same diseases.
- The study of the natural history of these conditions (longitudinal), the risk factors, severity and associated complications for example:
- cardiovascular complications
- gastro-intestinal complications
- bleeding complications
- oral and dental manifestations
- ophthalmologic manifestations
- genito-urinary manifestations
- muscular manifestations
- neurologic complications
- pregnancy-related problems manifestations
- mental health
- The identification of factors that could explain various phenotypes, including the studies of genotype/phenotype correlation.
- Quality of life studies.
At the moment, there is no cure for EDS or HSD. Management strategies are based on prevention and supportive treatment of symptoms, and, dependent on the underlying condition and observed clinical manifestations. At present there is no general consensus on the best practice for medical surveillance, management or surgical intervention of major complications, and there is a need for development of evidence-based recommendations for treatment in order to optimize medical care and improve patient health.
A major hurdle to the development of pharmacological and non-pharmacological treatment strategies, such as orthopedic and vascular surgery, physiotherapy, and pain management, is clinical trial design that can definitively determine whether a therapy is effective. Most EDS types are individually rare, and trials are by definition small and inevitably ‘underpowered’ statistically. In addition, the time to diagnosis is excessive, the clinical phenotypes of the different EDS types are variable, and the natural history is not well-documented.
Recruiting fairly homogeneous or similar patient populations is therefore difficult, and robust outcome measures are often lacking. Opportunities include creating patient registries with clinical and molecular data, increasing awareness to reduce the diagnostic delay, stimulating international collaboration to recruit larger patient cohorts or group, and improving trial design.
Research priorities
Study of treatment/management outcomes (e.g. orthopedic surgery, cardiovascular surgery, bisphosphonates, physiotherapy, pharmacological pain management, orthotics etc.)
- Intervention research: non-pharmacological management including treatment of related disorders and problems including but not limited to pain, fatigue, autonomic dysfunction, disability, psychosocial wellbeing, quality of life, and adherence to treatment.
- Studies regarding optimal methods for wound closure.
- Well-designed international pharmacological clinical trials in vEDS
The Ehlers-Danlos Society is particularly keen to better understand the costs of, and reasons for delays in diagnosis of EDS and HSD. Surveys over the last decade have repeatedly shown the time from presentation with symptoms to diagnosis is on average 12-14 years, and for some individuals decades. Costs include monetary impact (both at a personal individual level, and to health care systems, and economies) and effect on wellbeing.
Among the outcomes of good quality research in this area we envisage better understanding of the educational methods by which to inform both the public and health and social care professionals, and better understanding of what constitutes an effective health care service.

